Human iPS Cell Line (Amyotrophic Lateral Sclerosis)
- Specification
- Background
- Scientific Data
- Q & A
- Customer Review
Human iPS Cell Lines (iPSCs) derived from Amyotrophic Lateral Sclerosis (ALS) patients are generated by reprogramming somatic cells, such as dermal fibroblasts or peripheral blood mononuclear cells, obtained from individuals diagnosed with ALS. These cell lines retain the specific genetic background and disease-associated mutations (e.g., in SOD1, C9orf72, TARDBP, or FUS) of the donor. Characterized by standard pluripotency markers (e.g., OCT4, NANOG, SOX2, SSEA-4) and a normal karyotype, they exhibit indefinite self-renewal and the capacity to differentiate into all three germ layers.
In culture, they grow as adherent, tightly packed colonies with a high nuclear-to-cytoplasmic ratio, typically maintained in a nutrient-supplemented basal medium under standard conditions (37°C, 5% CO₂). Their primary value lies in disease modeling; they can be directed to differentiate into motor neurons and glial cells, allowing researchers to study ALS pathogenesis, protein aggregation, and neural degeneration in a patient-specific human context. Consequently, they are widely used for high-throughput drug screening and the development of targeted therapies for this fatal neurodegenerative disorder.
ATAC-Seq Data were Generated for 533 iPSC-Derived Motor Neuron Lines
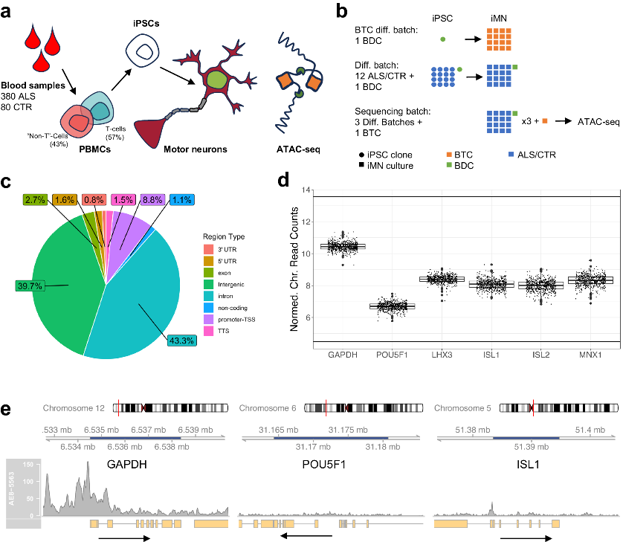
To investigate the epigenetic basis of sporadic ALS, Tsitkov et al. performed ATAC-seq on iPSC-derived motor neurons from 380 patients and 80 controls. ATAC-seq was conducted on 533 motor neuron lines from 460 donors, with variation controlled using batch differentiation controls (BDCs) and technical controls (BTCs) (Fig. 1a, b). All samples met ENCODE alignment quality standards. The consensus peak set comprised 100,363 regions, predominantly intronic/intergenic (80%) or promoter/5'UTR (10%) (Fig. 1c). Validation confirmed accessibility of motor neuron-specific genes (e.g., LHX3, ISL1) and silencing of the pluripotency marker POU5F1 (Fig. 1d), with high data reproducibility confirmed by inter-sample correlations.
Ask a Question
Write your own review
- Adipose Tissue-Derived Stem Cells
- Human Neurons
- Mouse Probe
- Whole Chromosome Painting Probes
- Hepatic Cells
- Renal Cells
- In Vitro ADME Kits
- Tissue Microarray
- Tissue Blocks
- Tissue Sections
- FFPE Cell Pellet
- Probe
- Centromere Probes
- Telomere Probes
- Satellite Enumeration Probes
- Subtelomere Specific Probes
- Bacterial Probes
- ISH/FISH Probes
- Exosome Isolation Kit
- Human Adult Stem Cells
- Mouse Stem Cells
- iPSCs
- Mouse Embryonic Stem Cells
- iPSC Differentiation Kits
- Mesenchymal Stem Cells
- Immortalized Human Cells
- Immortalized Murine Cells
- Cell Immortalization Kit
- Adipose Cells
- Cardiac Cells
- Dermal Cells
- Epidermal Cells
- Peripheral Blood Mononuclear Cells
- Umbilical Cord Cells
- Monkey Primary Cells
- Mouse Primary Cells
- Breast Tumor Cells
- Colorectal Tumor Cells
- Esophageal Tumor Cells
- Lung Tumor Cells
- Leukemia/Lymphoma/Myeloma Cells
- Ovarian Tumor Cells
- Pancreatic Tumor Cells
- Mouse Tumor Cells