Amyotrophic Lateral Sclerosis (ALS) Model
Creative Bioarray provides industry-standard animal models for Amyotrophic Lateral Sclerosis (ALS) research. The SOD1-G93A transgenic mouse model is a well-known preclinical platform for efficacy evaluation, mechanistic studies, and translational research, supported by rigorously validated study designs.

- Background
- Models
- Study Examples
- Features
- FAQ
Background
ALS is a progressive neurodegenerative disease primarily affecting upper and lower motor neurons, leading to progressive muscle weakness, impaired motor function, and eventual respiratory failure. ALS affects approximately 450,000 individuals worldwide. In North America and Europe, the incidence is 2 to 3 cases per 100,000 people each year, and the prevalence is 4 to 8 cases per 100,000 people. Reports from China and other Asian countries show that more people are getting the disease. Currently approved therapies, such as Riluzole and Edaravone, provide only limited clinical benefit by modestly slowing disease progression but do not prevent motor neuron degeneration.
ALS is characterized by a complex interplay of- cellular stress mechanisms and genetic anomalies. Mutations in SOD1, TDP-43, and C9orf72 cause protein misfolding, oxidative stress, mitochondrial dysfunction, impaired axonal transport, and excitotoxicity, culminating in selective motor neuron death. The breakdown of the neuromuscular junction (NMJ) leads to muscle atrophy, while the activation of microglia and inflammatory signals make neuronal damage worse. The SOD1 G93A mutation replicates critical features of familial ALS by specifically promoting the aggregation of misfolded SOD1 protein.
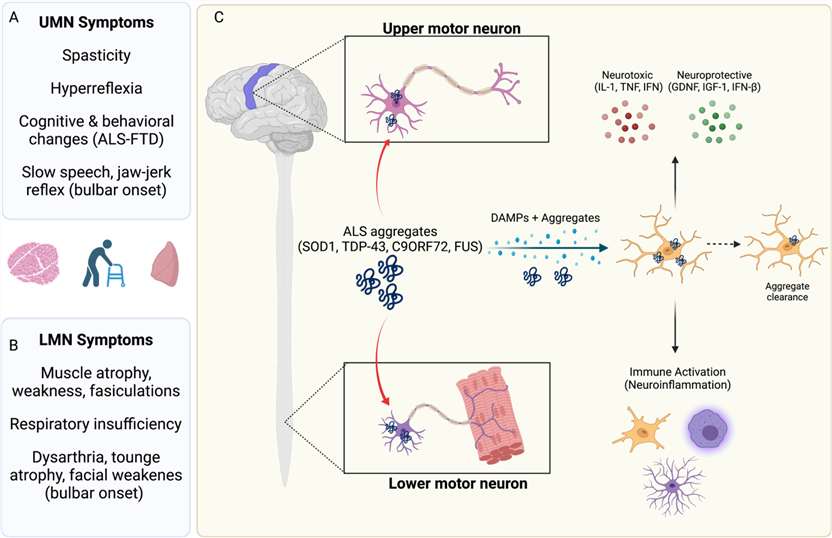 Fig. 1. Amyotrophic lateral sclerosis (ALS) symptoms and pathogenesis (Neel DV, Basu H, et al., 2022).
Fig. 1. Amyotrophic lateral sclerosis (ALS) symptoms and pathogenesis (Neel DV, Basu H, et al., 2022).
Because current therapies have limited efficacy, preclinical animal models are critical for investigating disease progression, testing mechanistic hypotheses, and evaluating prospective therapeutics in a controlled environment. The SOD1-G93A transgenic mouse is a reliable and reproducible model extensively utilized in ALS research.
Creative Bioarray's Amyotrophic Lateral Sclerosis (ALS) Model
The SOD1-G93A mouse has a human SOD1 gene with the G93A point mutation, which causes misfolded superoxide dismutase protein, progressive motor neuron degeneration, NMJ dysfunction, and muscular atrophy. This model accurately mimics numerous pathogenic and functional characteristics of human ALS, including gradual deterioration in motor performance and identifiable biochemical markers.
Animal Strain:
SOD1-G93A transgenic mouse model (C57BL/6J background)
Endpoints:
- Clinical outcomes: Body weight monitoring and survival analysis
- Behavioral and functional assessments: Rotarod test (motor coordination), grip strength (forelimb and hindlimb), gait analysis (stride length and cadence), and wire hang test (neuromuscular function)
- Pathology: Motor neuron quantification, NMJ morphology and skeletal muscle histology
- Biochemical analysis: Neurofilament light chain (NfL) in plasma and/or CSF, oxidative stress markers, and SOD1 aggregation
- Other customized endpoints: available upon request
Study Examples
Weight Loss and Motor Decline in SOD1 G93A Mice
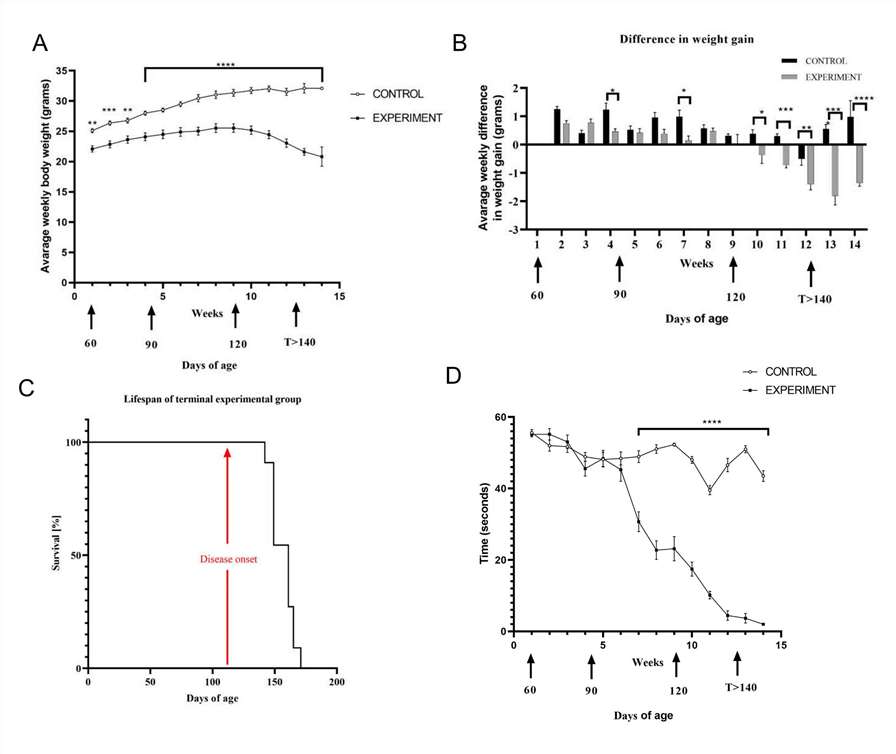 Fig. 2. SOD1-G93A mice develop progressive ALS-like phenotypes characterized by body weight loss, reduced motor performance, and shortened survival. (A-B) Body mass changes (C) Kaplan–Meier curves showing survival probability of experimental terminal group (D) Results of hanging cage test (Nowicka N, Szymańska K, et al., 2022).
Fig. 2. SOD1-G93A mice develop progressive ALS-like phenotypes characterized by body weight loss, reduced motor performance, and shortened survival. (A-B) Body mass changes (C) Kaplan–Meier curves showing survival probability of experimental terminal group (D) Results of hanging cage test (Nowicka N, Szymańska K, et al., 2022).
ASC-Exosomes Administration Improves Motor Performance of SOD1(G93A) Mice
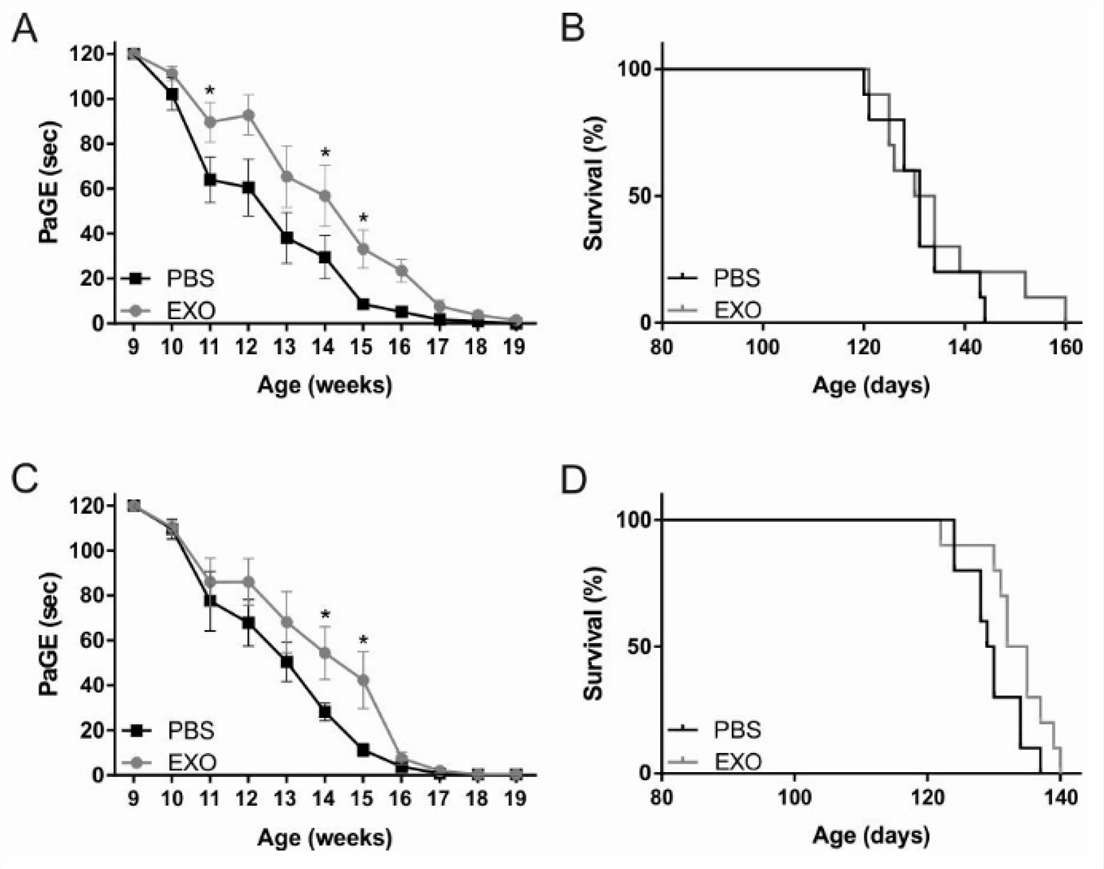 Fig. 3. Motor performances and survival of human SOD1 gene with a G93A mutation (SOD1(G93A)) mice treated intravenously (A, B) and intranasally (C, D) (Bonafede R, Turano E, et al., 2020).
Fig. 3. Motor performances and survival of human SOD1 gene with a G93A mutation (SOD1(G93A)) mice treated intravenously (A, B) and intranasally (C, D) (Bonafede R, Turano E, et al., 2020).
Why Choose Creative Bioarray for ALS Preclinical Research

In-Depth Knowledge of the ALS Model
Knowledgeable in disease staging, phenotype characterisation, and endpoint selection using the SOD1-G93A transgenic mice in accordance with established guidelines for ALS research.

Extensive Measurements for Function and Mechanism
A comprehensive evaluation is conducted that includes motor function tests (rotarod, grip strength, and wire hang test), survival analysis, tests for spinal cord disease (motor neuron loss and NMJ integrity), and circulating biomarkers to enable a multi-dimensional evaluation of efficacy.
Study Design Optimization Utilizing Disease Timeline
Adaptable study designs that take into account symptomatic vs. pre-symptomatic intervention, dosage regimens, duration of follow-up, and survival outcomes to accommodate various approaches to drug discovery.

Ensuring Consistent Quality and Reliability
To guarantee uniform and repeatable outcomes across experiments, researchers used standardized breeding, genotyping, randomization, and blinded assessments in conjunction with controlled experimental circumstances.
Frequently Asked Questions
What stages of the disease can be looked at?
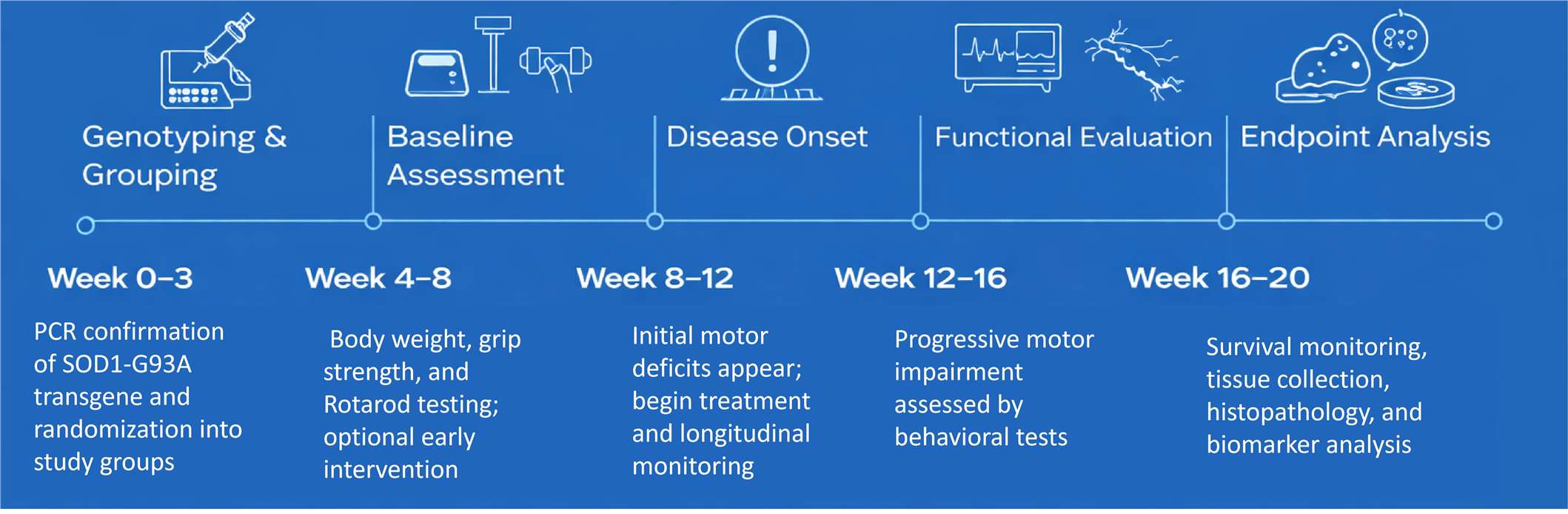
You can study both the pre-symptomatic (Weeks 4–8) and the symptomatic (Weeks 8–16) stages. This enables the assessment of preventive, disease-modifying, or symptomatic treatment strategies within a singular model.
What are the most important efficacy endpoints?
Common endpoints are changes in body weight, survival, spinal cord motor neuron counts, NMJ integrity, and biomarkers like NfL or oxidative stress markers. Other common endpoints are motor performance tests like the Rotarod, grip strength, and hanging test.
How do researchers make sure that their studies can be repeated?
Standardized transgenic colony management, PCR genotyping, random group allocation, blinded assessments, and consistent testing protocols all help keep reproducibility.
What kinds of drugs can be tested?
The model works with a lot of different types of treatments, such as small molecules, biologics, antisense oligonucleotides (ASOs), and gene therapies. It also lets you choose when and how to give them.
Accelerate your ALS drug discovery program with Creative Bioarray's SOD1-G93A transgenic mouse model and comprehensive in vivo preclinical CRO services. Contact us today to design scientifically robust studies for translational ALS research.
References
- Neel DV, Basu H, et al. Catching a killer: Mechanisms of programmed cell death and immune activation in Amyotrophic Lateral Sclerosis. Immunol Rev. 2022. 311(1):130-150.
- Nowicka N, Szymańska K, et al. The Involvement of RAGE and Its Ligands during Progression of ALS in SOD1 G93A Transgenic Mice. Int J Mol Sci. 2022. 23(4):2184.
- Bonafede R, Turano E, et al. ASC-Exosomes Ameliorate the Disease Progression in SOD1(G93A) Murine Model Underlining Their Potential Therapeutic Use in Human ALS. Int J Mol Sci. 2020. 21(10):3651.