
Human Skeletal Muscle Microvascular Endothelial Cells
Cat.No.: CSC-C4867L
Species: Human
Source: Skeletal Muscle
Cell Type: Endothelial Cell; Microvascular Cell
- Specification
- Background
- Scientific Data
- Q & A
- Customer Review
Never can cryopreserved cells be kept at -20 °C.
Human Skeletal Muscle Microvascular Endothelial Cells (HSMMECs) develop from the capillary networks within skeletal muscle tissue. Capillaries function as essential pathways for the exchange of materials and metabolic processes in skeletal muscle. Research shows that skeletal muscle capillaries have tight junctions similar to those found in the blood-brain barrier and blood-nerve barrier which help preserve both homeostasis and defense mechanisms in skeletal muscle tissue. HSMMECs function as capillary endothelial cells which deliver oxygen and nutrients including glucose along with metabolic products like lactate from blood to skeletal muscle cells.
The release of multiple cytokines and chemokines from these cells actively modulates immune system responses. For example, inflammatory factors like TNF-α and IL-1α boost ICAM-1 production in HSMMECs leading to increased leukocyte adhesion and migration. HSMMECs utilize tight junctions and paracellular pathways to protect skeletal muscle tissue by blocking harmful substances from penetrating the tissue. These cells perform essential functions in both skeletal muscle metabolism processes and signal transmission. Studies reveal that HSMMECs engage in inflammatory processes by producing inducible nitric oxide synthase (iNOS) while controlling endothelial cell morphology and functionality through redox signaling driven by NADPH oxidase. In vitro, HSMMECs can form vessel-like structures (tubules), indicating their angiogenic potential.
Endothelial ERΑ Promotes Endothelial Insulin Transport via Both Nuclear and Non-Nuclear Mechanisms
The estrogen receptor ERα plays a vital role in glucose homeostasis through its involvement in numerous glucose regulation processes across different tissues. The impact of ERα has been well-documented within the central nervous system, liver, skeletal muscle and adipose tissue but its function in endothelial cells has not yet been explored. Sacharidou et al. focused on analyzing the impact of ERα in endothelial cells on glucose regulation along with its potential role in boosting insulin transport to skeletal muscle.
Here they show that endothelial ERα promotes glucose control by enhancing muscle insulin delivery. How endothelial ERα positively influences skeletal muscle insulin delivery was then determined. Insulin delivery to skeletal muscle is limited by its transcytosis across the endothelial monolayer. They examined whether ERα affects insulin transport in endothelial cells. Research using human aortic endothelial cells (HAEC) demonstrated that estrogen (E2) enhances FITC-insulin uptake which can be stopped using both the ERα antagonist MPP and the PI3 kinase inhibitor LY294002 (Fig. 1a, b). The human skeletal muscle microvascular endothelial cells (HSMEC) displayed similar results when E2 enhanced insulin uptake through ERα and PI3 kinase pathways (Fig. 1c). Estrogen (E2) increased insulin transcytosis more than tenfold in HAEC and showed comparable effects in HSMEC (Fig. 1d-f). The results of Transwell assays showed stable monolayer barriers which demonstrated transcellular insulin transport mechanisms. In mouse skeletal muscle endothelial cells, E2 increased insulin uptake 3.2-fold in ERαfl/fl mice, which was blocked by PI3 kinase inhibition, but no increase was seen in ERαΔEC mice (Fig. 1g). These findings suggest a crucial role for ERα and PI3 kinase in insulin transport, linking in vivo and cell culture data.
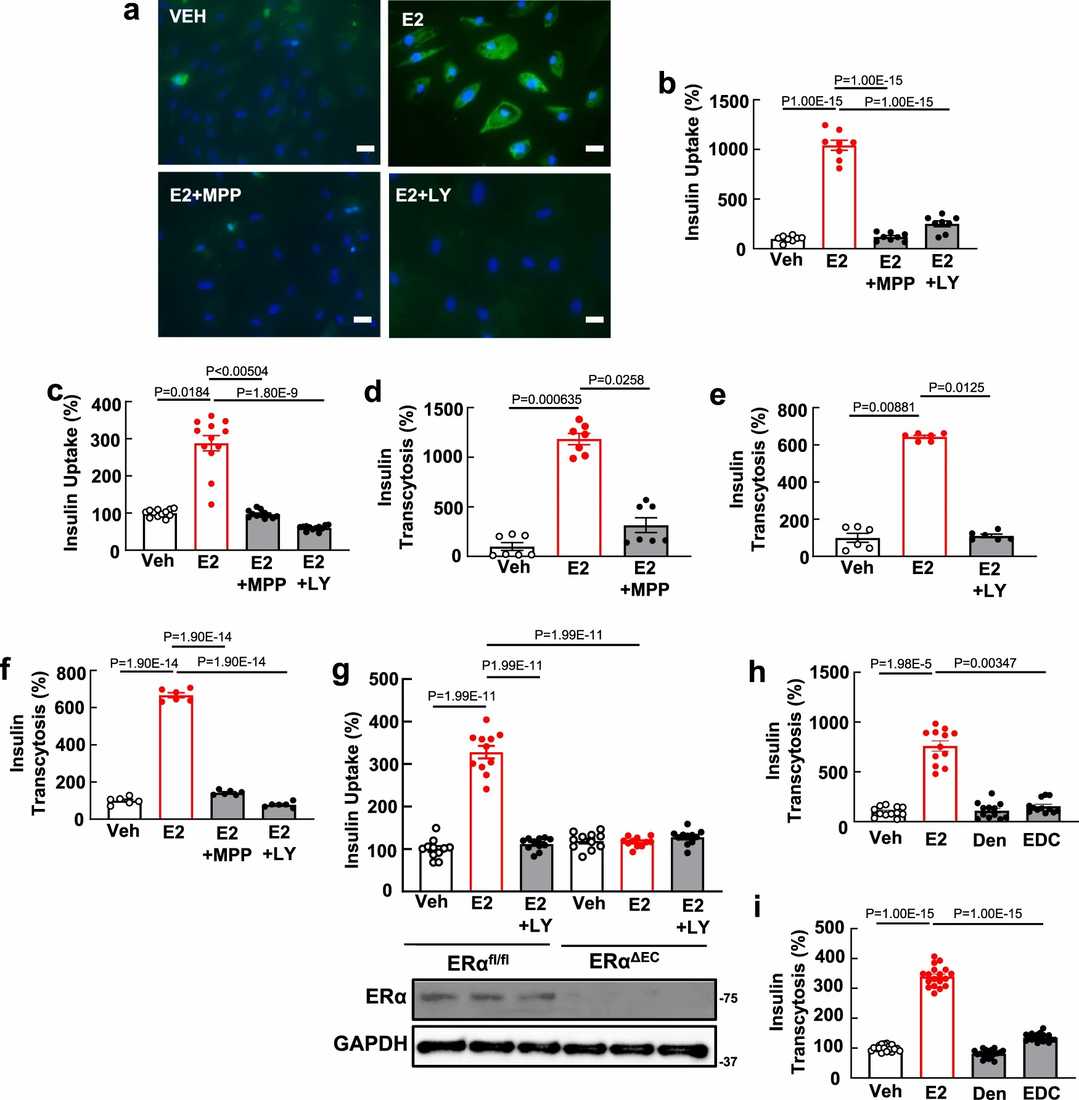 Fig. 1. Endothelial ERα promotes insulin transport by skeletal muscle microvascular endothelial cells via nuclear and non-nuclear actions of the receptor, with the latter entailing activation of PI3 kinase (Sacharidou A, Chambliss K, et al., 2023).
Fig. 1. Endothelial ERα promotes insulin transport by skeletal muscle microvascular endothelial cells via nuclear and non-nuclear actions of the receptor, with the latter entailing activation of PI3 kinase (Sacharidou A, Chambliss K, et al., 2023).
TRAF3IP2 Overexpression Impairs Insulin Signaling in Endothelial Cells and Blunts Endothelium-Dependent Relaxation in Isolated Arteries
Overweight individuals face increased risks of heart conditions due to connections between obesity and both insulin resistance and endothelial dysfunction which together drive vascular diseases. Grunewald et al. examined how TRAF3IP2 functions as a proinflammatory adaptor protein that influences vascular insulin resistance and dysfunction associated with obesity.
As displayed in Figure 2A, insulin treatment induced Akt activation (ie, Akt phosphorylation at Ser473/total Akt) in both control and TRAF3IP2-overexpressing cells (human skeletal muscle microvascular endothelial cells and human aortic endothelial cells); however, the level of Akt activation was significantly less in TRAF3IP2-overexpressing cells (P<0.05). Total Akt expression was not different between the treatments. Further, as shown in Figure 2B and 2C, adenoviral overexpression of TRAF3IP2 in isolated aortic rings blunted ACh-induced relaxation relative to control GFP expressing rings (P<0.05). In comparison, SNP-induced relaxation was not different between the treatment conditions (P>0.05). The suppression in ACh-induced aortic vasorelaxation caused by TRAF3IP2 overexpression was in alignment with the downregulation of eNOS in aortic endothelial cells (P<0.05), as displayed in Figure 2D.
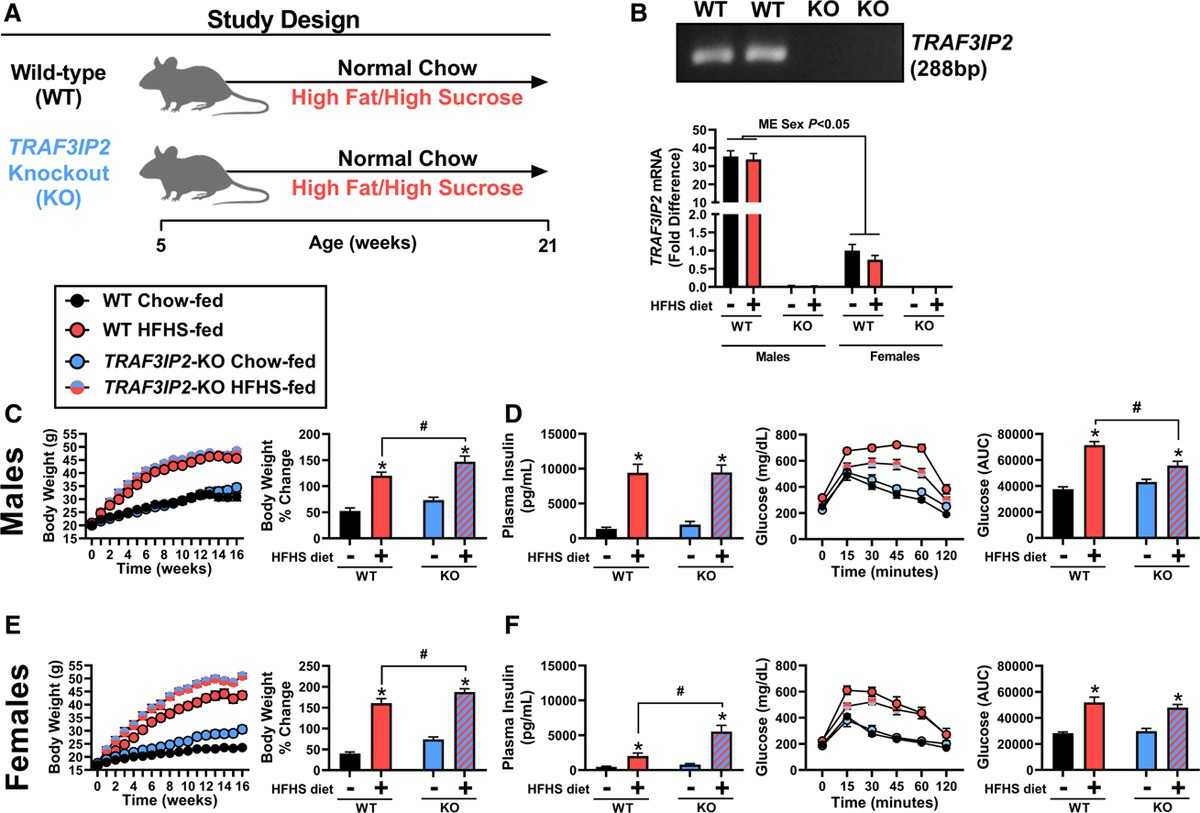 Fig. 2. RAF3IP2 deletion protects male mice against obesity-associated glucose intolerance (Grunewald ZL, Ramirez-Perez FL, et al., 2020).
Fig. 2. RAF3IP2 deletion protects male mice against obesity-associated glucose intolerance (Grunewald ZL, Ramirez-Perez FL, et al., 2020).
Ask a Question
Write your own review
Description: HNPC from Creative Bioarray are isolated from nucleus pulposus of human intervertibral disc. HNPC are cryopreserved at primary or passage one culture and delivered frozen. Each vial contains >5 x ...
Description: HS from Creative Bioarray Research Laboratories are isolated from human synovium. HS are cryopreserved at passage one cultures and delivered frozen. Each vial contains >5 x 10^5 cells in 1 ml ...
Description: HC-a from Creative Bioarray are isolated from human articular cartilage. HC-a are cryopreserved at primary cultures and delivered frozen. Each vial contains >5 x 10^5 cells in 1 ml volume. HC-a are ...
Description: Description: Human skeletal muscle cells are derived from whole muscle that has been dissociated into single cells and frozen. These muscle cells are from a single donor. They enable researchers to ...
Description: Human Tenocytes are isolated from the patellar or Achilles tendon. The cells are isolated either by centrifugal force following enzymatic treatment or from an explants culture. Human Tenocytes are a ...
Description: HAFC from Creative Bioarray are isolated from annulus fibrosus of human intervertibral disc. HAFC are cryopreserved at primary or passage one culture and delivered frozen. Each vial contains >5 x ...