LATS Kinase Phosphorylation Assay by Immunoblotting
This assay describes the immunodetection of phosphorylated proteins by immunoblot analysis after detergent extraction. Phosphorylation of large tumor suppressor 1/2 (LATS1/2) is examplified, however, this protocol can also be used for most specific anti-phosphoamino acid or anti-phosphoprotein antibodies.
Large tumor suppressor 1/2 (LATS1/2) are protein kinases and core components of the Hippo pathway, which regulate organ size and tissue homeostasis. LATS kinase is activated by phosphorylation on its hydrophobic motif (HM, Thr1079 for LATS1 and Thr1041 for LATS2). Western blotting with phosphoantibody to recognize LATS at HM provides an indirect way to assess LATS kinase activity.
This protocol minimizes the time phosphorylated proteins are exposed to phosphatases, allowing reliable and quantitative detection of the phosphorylated proteins, and can be completed within 7 to 10 hours.
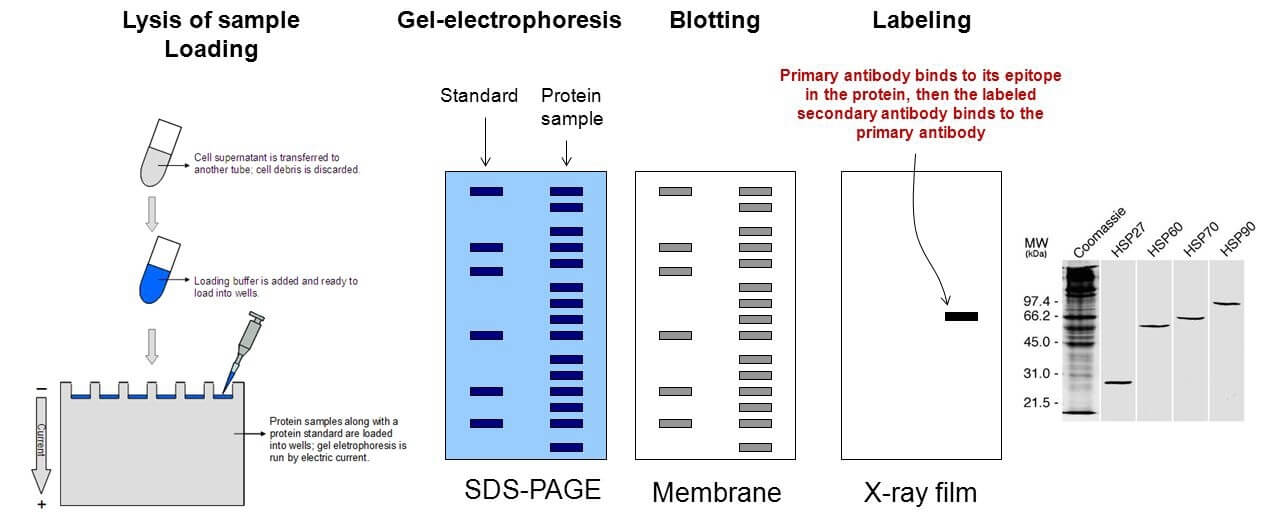 Figure 1. The general workflow for immunoblotting.
Figure 1. The general workflow for immunoblotting.
Materials
Table 1. List of required materials.
| Conventional medium (DMEM/50% FBS) | Starvation medium (DMEM/1% FBS) |
| Bovine serum albumin (BSA) | Phosphate buffered saline (PBS) |
| Epidermal growth factor (EGF) 50 μg/mL | EGF buffer (PBS/0.5 mg/mL BSA) |
| Cell lines of interest (primary murine karatinocytes) | Protein standards (5, 10, 20, 50, 100, and 200 μg/mL BSA in homogenization buffer) |
| Coomassie protein assay reagent | Prestained protein markers |
| 12% SDS-polyacrylamide gel (12% polyacrylamide/0.32% bisacrylamide) | Lysis buffer (homogenization buffer/1% Triton X-100) |
| Transfer buffer (50 mM Tris-HCl/50 mM glycine) | Blocking solution (TBST/2% BSA) |
| Primary antibody (monoclonal anti-active LATS kinase) | Secondary antibody (horseradish peroxidase (HRP)-conjugated) |
Equipment
- Enhanced chemiluminescence (ECL) detection system
- Microtiter plate reader
- Microfuge tubes
- Centrifuge
- Nitrocellulose membrane
- Transfer apparatus for electroblots
- Flat container for washing nitrocellulose membrane
Reagent Preparation
- Homogenization buffer
50 mM β-glycerophosphate (pH 7.3), 1.5 mM EGTA, 1.0 mM EDTA, 0.1 mM sodium vanadate, 1.0 mM benzamidine, 10 μg/mL leupeptin, 2.0 μg/mL pepstatin A, 1.0 mM dithiothreitol (DTT), store up to 3 months at 4°C without DTT, add DTT just before use. - Kinase buffer
50 mM β-glycerophosphate (pH 7.3), 1.5 mM EGTA, 1.0 mM EDTA, 0.1 mM sodium vanadate, 1.0 mM DTT, store up to 3 months at 4°C without DTT, add DTT just before use. - 4X SDS-polyacrylamide gel electrophoresis (PAGE) sample buffer
200 mM Tris-HCl (pH 6.8), 40% (v/v) glycerol, 8% (w/v) SDS, 0.2% (w/v) bromphenol blue, 8% (v/v) 2-mercaptoethanol, store up to 12 months at -20°C. - Tris-buffered saline/Tween 20 (TBST)
20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.05% (v/v) Tween 20, store up to 3 months at 4°C.
Assay Procedure
Cell extract preparation
- Plate cell lines of interest in DMEM medium with 10% FBS. Incubate at 37°C, 5% CO2 to reach a confluency of 80%.
- Replace the conventional culture medium with starvation medium and further culture for 18 h.
Note: Serum starvation makes the cells quiescent, which can be achieved within 14 to 24 h. Starvation for too long or any change in temperature or pH may be stressful to the cells and may induce activation of one or more signaling pathways. - Add 2.5 μL of 50 μg/mL EGF to three plates (stimulated) and 2.5 μL EGF buffer to three plates (control). Return the plates to the incubator.
Note: It is necessary to ensure that stimulus is given at the appropriate time and the cells harvested within a short period of time (5-10 min). 2) If the influence of the stimulant agent on the particular cells being used is not yet known, a positive control should be included. For example, peroxovanadate (0.1 mM vanadate/0.2 mM hydrogen peroxide), which nonspecifically activates many signaling events in most tissue culture cells. - After 5, 15, or 45 min, remove starvation medium from the plates. Rinse twice with 5 mL ice-cold PBS and once with 5 mL ice-cold homogenization buffer.
- Add ice-cold lysis buffer to each plate, tilt the plate gently and scrape the cells into the buffer using a plastic scraper. Transfer the cells and buffer to pre-labeled, pre-chilled 1.5 mL microfuge tubes.
Note: The sample being harvested may become slightly viscous, so the 1 mL pipet tips can be cut to allow easy collection of these samples. 2) The cell lysate should be harvested within 10 min. 3) Detergents other than Triton X-100, such as 0.5% nonidet P-40 (NP-40), can also be used. - Centrifuge the cellular extracts at 15,000 x g for 15 min at 4°C. Transfer supernatants to fresh, pre-chilled, and pre-labeled microfuge tubes.
Note: The supernatants contain the protein extracts to be examined for phosphorylation. - Transfer 5-10 μL aliquots of each extract to labeled microfuge tubes for determination of protein concentration, and store the remainder of the extracts on ice.
Note: The protein concentration is determined at this stage so that identical amounts of proteins from the different samples can be compared and the relative amount of phosphor-proteins can be determined accurately.
Protein concentration determination
- Dilute the cell extracts by add 145 μL of kinase buffer to each 5 μL of cell extract.
- Transfer 10 μL of each of the protein standards into two wells of a 96-well plate.
Note: Protein standards should be prepared in the same buffer as was used for cell extraction. - Transfer 10 μL of each of the diluted cellular extracts into two well of the same plate. Add 200 μL coomassie protein assay reagent to all wells.
- Place the 96-well plate in a microtiter plate reader and measure the absorbance of the standards and samples at 595 nm. Use the absorbance of the standards to construct a standard curve. Calculate the protein concentrations of the samples by comparing the absorbance of the samples with the standard curve.
SDS-PAGE analysis
- Based on the determined protein concentrations, transfer each cellular extract containing 40 μg of protein to a fresh 1.5 mL microfuge tube.
- Add 1/3 vol. of 4X SDS-PAGE sample buffer to each tube, mix and boil for 3 min.
- Load samples and pre-stained protein markers on 12% SDS-polyacrylamide gel.
Note: Load pre-stained markers into the first or second lane of the gel so that the molecular weights of the detected proteins can be determined. These markers will also indicate whether the proteins are completely transferred from the gel onto the nitrocellulose paper during blotting. - Place the gel in an electrophoresis apparatus filled with appropriate buffer and run the gel at 150 V.
Protein immunoblotting
- Soak nitrocellulose membrane in transfer buffer until completely wet.
- Once the dye front has reached the end of the gel, remove the gel from the electrophoresis apparatus, cut off the stacking (upper) gel, and carefully place the gel in a flat container with transfer buffer.
- Fill transfer apparatus with transfer buffer. Open the inner transfer apparatus and remove air bubbles from the pads. Make a sandwich of the gel, nitrocellulose membrane, and pads by putting one piece of 3 mm filter paper (wetted with transfer buffer) on the wet pad, the gel on top of the filter paper, the wet nitrocellulose membrane on top of the gel, and the second piece of wet 3 mm paper on top of the nitrocellulose membrane.
- Remove any air bubbles from between the different layers of the transfer sandwich by gently rolling a 10 mL pipet over the sandwich. Place the other wet pad on top of the transfer sandwich.
Note: Make sure air bubbles are not trapped between the gel and the other components. - Place the transfer sandwich containing the gel and nitrocellulose membrane into the buffer-filled transfer apparatus, with the nitrocellulose membrane facing the side with the cathode and the gel facing the side with the anode. Connect the apparatus to a power supply and start the current (200 mA constant current, preferably with a cooling device). Run for 2 h.
Note: To shorten the time and improve the yield of transfer, methanol or 0.05% SDS is sometimes included in the transfer buffer, their inclusion will allow higher current but will necessitate the use of a cooling device. - Turn off the power supply and remove the nitrocellulose membrane from the transfer sandwich. Rinse the nitrocellulose membrane with transfer buffer to remove any adhering pieces of gel and place the membrane in a flat container.
- Place nitrocellulose membrane into flat container and incubate in 30-50 mL of blocking solution for 60 min at room temperature.
Note: This ensures that any free nonspecific protein-binding sites on the membrane are blocked and will not nonspecifically bind the antibodies to be used.
Incubating nitrocellulose membrane with antibody
- Dilute primary antibody in TBST according to manufacturer’s instructions. Incubate nitrocellulose membrane in a flat container with 15 mL of primary antibody overnight at 4°C, 30 min at 37°C, or 1-2 h at room temperature.
- Wash nitrocellulose membrane with TBST in flat container at least three times, 15 min each time.
- Dilute secondary antibody in TBST according to manufacturer’s instructions. Incubate nitrocellulose membrane with secondary antibody for 45 min at room temperature.
- Wash nitrocellulose membrane with TBST at least three times, 10 min each time.
- Incubate nitrocellulose membrane with ECL solution for 1 min, dry the blot with filter paper, wrap the blot in plastic wrap, and expose to X-ray film.
Cell Services
Cell Line Testing and Assays