Chromosome Conformation Capture
Chromosome conformation capture (3C) is a technique for detecting the spatial organization of chromosomal DNA in fixed cells. 3C can be used to study long-range interactions between DNA sequences on the same chromosome (intra-chromosomal) or on different chromosomes (inter-chromosomal). Briefly, cells are fixed with formaldehyde, which forms DNA-protein and protein-protein cross-links between regions of the genome in proximity. Subsequent restriction enzyme digestion and intra-molecular ligation produce novel junctions between restriction fragments in the nucleus. Polymerase chain reaction (PCR) is then used to amplify the DNA fragments containing novel ligation junctions using primers specific for a pair of DNA sequences.
3C technique plays a key role in investigating how nuclear organization and structural interactions between regulatory elements relate to gene activity. As these interactions are highly tissue-specific, it is crucial that 3C experiments are performed in well defined, purified cell populations.
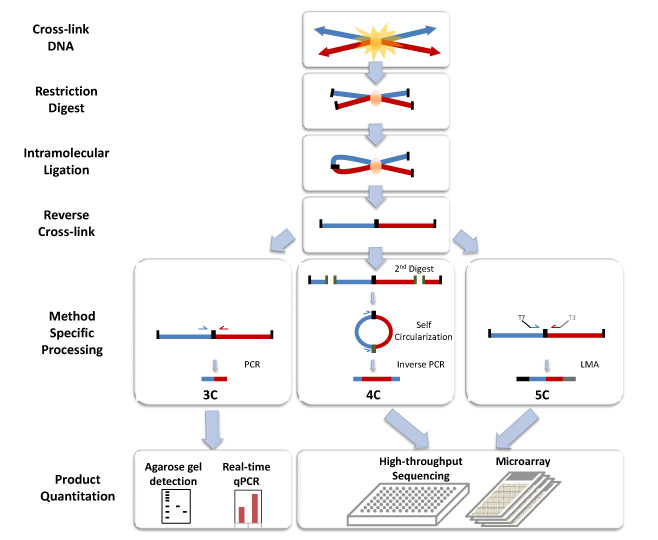 Figure 1. Chromosome conformation capture technology.
Figure 1. Chromosome conformation capture technology.
Materials and Equipment
| Agarose gel (2%) | Isopropanol | Ethidium bromide |
| Chloroform | Lysis buffer | Sodium acetate (2 M) |
| Control template | PCR primers | Water bath |
| SDS (Sodium dodecyl sulfate; 20%) | Phenol-chloroform-isoamyl alcohol (25:24:1) | Phosphate buffered saline (PBS) |
| Formaldehyde solution | T4 DNA ligase | TaqMan probe |
| HotStar Taq DNA polymerase | Quant-iT PicoGreen dsDNA assay kit | TaqMan universal PCR master mix |
| HindIII restriction enzyme | Proteinase K (10 mg/mL) | Tissue or cell type of interest |
| Glycine (1M) | RNase A (100 mg/mL) | Triton X-100 (20%) |
| Vortex mixer | Incubator | Centrifuge |
| SYBR green PCR master mix | Sequence detection system or equivalent | Agarose gel electrophoresis equipment |
Fixation
- Collect cells from tissue and make single-cell suspensions of 104-106 cells in 1 mL growth media.
- Add formaldehyde to a final concentration of 2%, mix well and incubate for 10 min at RT on a rocking or tumbling incubator.
- Quench by adding 150 μL 1 M cold glycine.
- Centrifuge at 500 g for 15 min at 4°C.
- Carefully remove supernatant, do not disturb pellet.
- Wash pellet by gently resuspending in 1 mL cold PBS.
- Centrifuge at 500 g for 15 min at 4°C.
- Carefully remove supernatant, do not disturb pellet.
- Resuspend pellet in 500 μL cold lysis buffer.
- Incubate for 20 min on ice.
- Collect the nuclei by centrifugation at 800 g for 6 min at 4°C.
Digestion
- Resuspend the collected nuclei in 1 mL of cold 1.2X NEBuffer 2.
- Count the nuclei using a hemacytometer. Prepare aliquots of 107 nuclei in 500 µL of 1.2X NEBuffer 2.
- Add 20% SDS (to a final concentration of 0.3%). Incubate with shaking at 950 rpm in a Thermomixer for 1 h at 37°C.
- Add 20% Triton X-100 (to a final concentration of 1.8%). Incubate with shaking at 950 rpm for 1 h at 37°C.
- Add 1500 U of HindIII. Digest with shaking at 950 rpm overnight at 37°C.
Ligation
- Add 20% SDS (to a final concentration of 1.6%). Incubate for 25 min at 65°C to inactivate the restriction enzyme.
- Transfer the sample to a 15 mL centrifuge tube containing 7 mL of 1.1X T4 DNA ligase reaction buffer.
- Add 20% Triton X-100 (to a final concentration of 1%). Incubate for 1 h at 37°C, mixing occasionally.
- Add 800 U of T4 DNA ligase. Incubate for 4 h in a 16°C water bath.
- Incubate for 30 min at room temperature.
De-crosslinking
- Add proteinase K to a final concentration of 100 µg/mL. Incubate at 65°C overnight.
DNA Purification
- Cool the samples to room temperature. Treat with RNase A (final concentration of 0.5 µg/mL) for 30 min at 37°C.
- Transfer each sample to a 50 mL centrifuge tube. Add 10 mL of phenol-chloroform-isoamyl alcohol and vortex thoroughly. Centrifuge at 2500 g for 15 min at room temperature.
- Transfer the aqueous phase to a clean 50 mL centrifuge tube. Add 7 mL of chloroform to the aqueous phase and vortex. Centrifuge at 2500 g for 15 min at room temperature.
- Carefully collect the aqueous phase. Add 2 M sodium acetate (to a final concentration of 0.8 M) to it. Precipitate DNA with 100% ethanol for 1-3 h at -20°C.
- Pellet the precipitated DNA at 2500 g for 30 min at 4°C.
- Wash the pelleted DNA with 20 mL of 70% ethanol. Centrifuge at 2500 g for 30 min at 4°C.
- Air-dry the pellet. Resuspend in 100 µL H2O.
- Pipette the samples vigorously. Incubate overnight at 37°C to fully resuspend the DNA.
- Quantify the 3C material using a PicoGreen dsDNA quantitation kit according to the manufacturer’s instructions.
Semiquantitative PCR Amplification
- Amplify unique 3C products by using HotStar Taq DNA polymerase and 250-300 ng DNA per PCR reaction with the following program:
- 15 min at 95°C
- 35 cycles of 1 min at 60°C, 1 min at 72°C, 30 sec at 94°C
- 2 min at 60°C
- 10 min at 72°C
- Hold indefinitely at 4°C
- Separate the PCR products on a 2% agarose gel. Stain with ethidium bromide.
Note: To ensure that primer efficiency does not introduce bias, include a control template that contains all possible ligation products in equimolar amounts. The amount of genomic DNA should mimic the amount of DNA used in the actual 3C samples (i.e., ~300 ng/reaction).
Quantitative PCR Amplification
- For SYBR Green PCR
- Mix 50 ng DNA from unique 3C products with primer pairs (100-400 nM) and 1X SYBR green PCR master mix in a final reaction volume of 25 µL.
- Assess ligation products relative to a control gene using a sequence detection system and the following program:
| Number of cycles | Temperature | Time |
| 40 | 50°C | 2 min |
| 95°C | 10 min | |
| 95°C | 15 sec | |
| 60°C | 1 min |
- For TaqMan PCR
- Mix 200-250 ng DNA from unique 3C products with TaqMan probes, primer pairs, and TaqMan universal PCR master mix (1X final concentration) in a final reaction volume of 25 µL.
- Assess ligation products relative to a control gene using a sequence detection system and the following program:
| Number of cycles | Temperature | Time |
| 44 | 50°C | 2 min |
| 95°C | 10 min | |
| 95°C | 15 sec | |
| 60°C | 1.5 min |
Note: Perform all reactions in triplicate. Compare against a control template standard curve to account for differences in primer pair efficiency. The primer concentrations should be optimized for each sample.
References
- Dekker J. et al.; Capturing chromosome conformation. Science, 2002, 295: 1306-1311.
- Cope N F. et al.; Chromosome conformation capture. Cold Spring Harb Protoc, 2009.
Cell Services:
Cell Line Testing and Assays