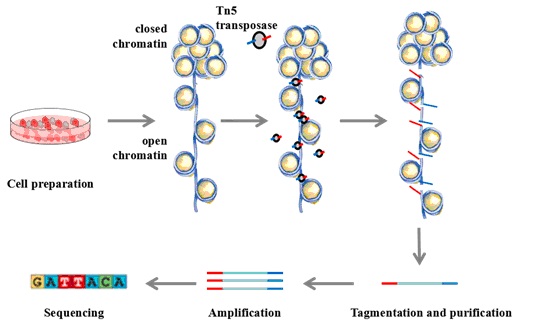
Open Chromatin Analysis
A number of publications have described methods for mapping open chromatin in a single cell. Here we introduce a method based on the analysis of transposase-accessible chromatin (ATAC-seq), which uses a Tn5 transposase enzyme to simultaneously fragment DNA and attach adapter sequences in a process called tagmentation. ATAC-seq is compatible with a wide variety of methods for cell separation and isolation including cell sorting and disruption of tissues to cell suspensions. This method also allows for adaptation to different cell types and species. Typically, ATAC-seq is separated into three separate components: cell lysis, transposition, and amplification. Cross-linking usually reduces library creation efficiency, so we recommend starting with fresh unfixed cells to maximize sensitivity.
Materials
- Phosphate buffered saline (PBS)
- Molecular biology grade IGEPAL
- Lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL)
- 2X TD (2X reaction buffer)
- TDE1 (Tn5 transposase)
- PCR Purification Kit
- High-fidelity 2X PCR Master Mix
- 25 uM Custom PCR Primer 1
- 25 uM Custom PCR Primer 2
- 100X SYBR Green I
- 0.2 mL PCR tubes
- PCR Thermal cycler
Cell Preparation
- Harvest cells, protocol to be defined by the user.
Note: Cells should be intact and in a homogenous single cell suspension. - Spin down 50,000 cells at 500 g for 5 min, 4°C.
- Wash once with 50 μL of cold 1X PBS buffer. Spin down at 500 g for 5 min, 4°C.
- Gently pipette to resuspend the cell pellet in 50 μL of cold lysis buffer. Spin down immediately at 500 g for 10 min, 4°C.
Note: This step provides lysis of cells with non-ionic detergent and generates of a crude nuclei preparation. - Discard the supernatant, and immediately continue to transposition reaction.
Transposition Reaction and Purification
- Make sure the cell pellet is set on ice.
- To make the transposition reaction mix, combine the following:
1) 25 μL TD (2X reaction buffer)
2) 2.5 μL TDE1 (Tn5 Transposase)
3) 22.5 μL Nuclease Free H2O - Resuspend nuclei in the transposition reaction mix.
- Incubate the transposition reaction at 37°C for 30 min.
Note: Gentle mixing may increase fragment yield. - Immediately following transposition, purify using a PCR Purification Kit.
- Elute transposed DNA in 10 μL Elution Buffer (10 mM Tris buffer, pH 8).
- Purified DNA can be stored at -20°C.
Note: This is a convenient stopping point. Please note that these DNA fragments are not PCR amplifiable if melted at this point.
PCR Amplification
- To amplify transposed DNA fragments, combine the following in a 0.2 mL PCR tube:
Note: Care should be taken to ensure that samples are barcoded appropriately for subsequent pooling and sequencing. - 10 μL Transposed DNA
- 10 μL Nuclease Free H2O
- 2.5 μL 25 μM Custom PCR Primer 1
- 2.5 μL 25 μM Custom PCR Primer 2 (contains barcode)
- 25 μL High-fidelity 2X PCR Master Mix
- Thermal cycle as follows:
Note: This first 5 minute extension at 72°C is critical to allow extension of both ends of the primer after transposition, thereby generating amplifiable fragments. This short pre-amplification step ensures that downstream quantitative PCR (qPCR) quantification will not change the complexity of the original library. - 1 cycle of 72°C for 5 min, 98°C for 30 sec
- 5 cycles of 98°C for 10 sec, 63°C for 30 sec, 72°C for 1 min
- To reduce GC and size bias in PCR, the appropriate number of PCR cycles is determined using qPCR, allowing us to stop amplification prior to saturation. To run a qPCR side reaction, combine the following in qPCR compatible consumables:
- 5 μL of previously PCR amplified DNA
- 4.41 μL Nuclease Free H2O
- 0.25 μL 25 μM Customized PCR Primer 1
- 0.25 μL 25 μM Customized PCR Primer 2
- 0.09 μL 100X SYBR Green I
- 5 μL High-fidelity 2X PCR Master Mix
- Using a qPCR instrument, cycle as follows:
- 1 cycle of 98°C for 30 sec
- 20 cycles of 98°C for 10 sec, 63°C for 30 sec, 72°C for 1 min
To calculate the additional number of cycles needed, plot linear Rn versus cycle and determine the cycle number that corresponds to ¼ of maximum fluorescent intensity.
Note: The purpose of this qPCR step is to generate libraries that are minimally PCR amplified. Most PCR bias comes from later PCR cycles that occur during limited reagent concentrations. This determination of the optimal number of cycles to amplify the library reduces artifacts associated with saturation PCR of complex libraries.- Run the remaining 45 μL PCR reaction to the cycle number determined by qPCR. Cycle as follows:
Note: Cycle for an additional N cycles, where N is determined using qPCR. - 1 cycle of 98°C for 30 sec
- N cycles of 98°C for 10 sec, 63°C for 30 sec, 72°C for 1 min
- Purify amplified library using PCR Purification Kit. Elute the purified library in 20 μL Elution Buffer (10 mM Tris Buffer, pH 8). Be sure to dry the column before adding elution buffer.
Note: The concentration of DNA eluted from the column ought to be approximately 30 nM, however 5-fold variation is possible and not detrimental.
Optional Library Quality Control
Prior to purification, the amplified libraries can be visualized by gel electrophoresis, however, due to the low concentration of the amplified materials, a 5% TBE polyacrylamide gel is required to optimize sensitivity. We found that adding 0.6X SYBR Green I to the libraries provides an excellent signal-to-noise without the need for post-staining. We usually load 15 ng of 100 bp NEB ladder with 0.6X SYBR Green I as a DNA marker. In principle, any instrument containing a blue-light source or imaging systems equipped with a laser that emits at 488 nm can be used to visualize DNA stained with SYBR Green I dye.
The quality of purified libraries can also be assessed using a Bioanalyzer High Sensitivity DNA Analysis Kit. We have seen that a preponderance of high molecular weight DNA (>1000 bp) can create misleading and inaccurate Bioanalyzer results.
References
- Thurman R E. et al.; The accessible chromatin landscape of the human genome. Nature, 2012, 489: 75-82.
- Buenrostro J. et al.; ATAT-seq: a method for assaying chromatin accessibility genome-wide. Current Protocols in Molecular Biology, 2015, 109: 21.29.1-21.29.9.
Cell Services
Cell Line Testing and Assays