DNA Methylation Profiling
DNA methylation, most commonly occurring in the C5 position of cytosines within CpG dinucleotides, plays a key role in many biological processes such as gene expression, embryonic development, cellular proliferation, differentiation, and chromosome stability. Abnormal DNA methylation is often associated with the loss of DNA homeostasis and genomic instability, which lead to the development of human diseases.
Bisulfite sequencing technology is considered to be the “gold standard” method for detecting DNA methylation because it provides a qualitative, quantitative and efficient approach to identify 5-methylcytosine at single base-pair resolution. This method is based on the amination reactions of cytosine and 5-methylcytosine (5mC) after the treatment of sodium bisulfite, which not only recognizes the methylation status along the DNA single strand, but also identifies the methylation patterns of DNA double strands since the converted DNA strands are no longer self-complementary and the amplification products can be measured individually. Bisulfite-based DNA methylation analysis are characterized by superior accuracy, high detection sensitivity, great efficiency, as well as a wide spectrum for sample analysis.
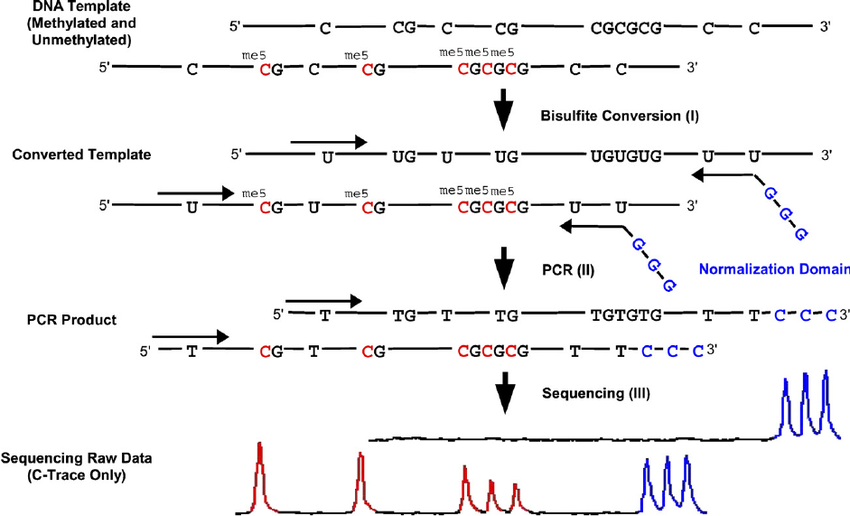 Figure 1. The principle of bisulfite-based DNA methylation analysis.
Figure 1. The principle of bisulfite-based DNA methylation analysis.
Materials
DNA Extraction Kit
Wizard DNA Clean-up Kit
Bisulfite Reaction Kit
70% of ethanol
Regular PCR Master Mix
PCR Purification Kit
Gel Extraction Kit
pGEM-T Easy Vector System II
Spin Miniprep Kit
Deionized water or TE buffer
Incubator
ABI 3730 DNA Analyzer
Disposable 5 mL lure-lock syringes
Microcentrifuge
Genomic DNA Preparation
Genomic DNA from cultured cells, cultured bacteria and animal tissues can be isolated by using a number of commercially-available DNA Extraction Kit followed by the corresponding manufacturer’s protocols. Genomic DNA (1-10 μg) is dissolved in deionized water with a final volume of 18 μL.
Note: The quality and quantity of DNA are important in the bisulfite reaction. In general, most protocols recommend >1 μg of high-quality DNA samples to obtain reliable results.
Bisulfite Modification
- Pre-denature the DNA by boiling in a water bath for 20 min.
- Denature the DNA by adding 2 μL of 3 M freshly made NaOH and 380 μL 5 M sodium bisulfite solution and mix well.
Note: The most critical step for the bisulfite reaction is DNA denaturation since sodium bisulfite can only react with cytosine in single-stranded DNA. Therefore, complete DNA denaturation is an essential prerequisite for successful DNA conversion by bisulfite treatment. Various modifications have been attempted to reduce strand re-annealing:1) DNA can be fragmented by the use of proteinase K and appropriate restriction enzymes. 2) DNA can be imbedded in low-melting-point agarose block to prevent DNA re-annealing. 3) DNA can be pre-boiled prior to bisulfite treatment to improve the denaturing step. 4) A high concentration of urea (6 M) may be added to the bisulfite solution to destabilize base-pairing in the DNA. - Add 500 μL of heavy mineral oil on the top of 400 μL DNA solution to diminish evaporation and incubate the solution in the dark at 50°C for 12-16 h.
- Purification of the bisulfite treated-DNA: We routinely use the Wizard DNA Clean-up Kit to purify the bisulfite treated-DNA. After carefully removing the heavy mineral oil from the reaction solution, the procedure involving this step is followed according to the manufacturer’s protocol.
- The bisulfite-modified DNA is eluted in 50 μL deionized water and 11 μL 3 M NaOH is added. Incubate at 37°C for 15 min to desulfonate the DNA.
- Add 166 μL 5 M ammonium acetate, 750 μL of absolute ethanol and 200 μL isopropanol to precipitate the DNA at -20°C for 2-4 h.
- Centrifuge the DNA at maximum speed for 10 min and discharge the supernatant.
- Wash the DNA with 200 μL 70% ethanol and centrifuge for 10 min.
- Carefully remove the ethanol and dry the DNA pellet at room temperature for 10 min. Resuspend the DNA in 10-20 μL of TE or deionized water.
Note: Owing to a non-complementary DNA conformation after bisulfite treatment, the converted DNA is not stable and repeated freeze-thawing should be avoided.
Bisulfite PCR Amplification
- The principles for designing bisulfite PCR primers vary to meet different research purposes. The primer guidelines listed below are used for bisulfite genomic sequencing analysis.
- After bisulfite treatment, the un-methylated cytosines are converted to thymine, while methylated cytosines remain cytosines. Since the methylation status of CpG dinucleotides is unknown, the bisulfite primer sequences should strictly avoid the CpG dinucleotides. Therefore, primers should be generated to replace all cytosines to thymines based on the original DNA sequence. Primer designing software can also be used to avoid potential hairpin structures and possible primer dimers based on this modified sequence.
- Primer length should be around 25 to 30 nucleotides.
- The length of the PCR product should not exceed 400 bp, as potential DNA degradation during the bisulfite modification may affect the PCR amplification.
Note: Bisulfite PCR primer design is critical for successful implementation of subsequent bisulfite sequencing analysis. - Bisulfite PCR amplification can be performed as a conventional PCR reaction. However, the PCR conditions should be carefully optimized because bisulfite treatment reduces the specificity of DNA duplex.
- Annealing temperature: A gradient PCR thermocycler can help to determine the appropriate annealing temperature. If there is no access, a touchdown PCR can also be applied to increase the annealing sensitivity.
- PCR reaction system: The commercially-available PCR Master Mix combining Taq DNA polymerase and dNTP with optimal salt concentration can be easily used for bisulfite PCR. If this common PCR reaction system does not produce clean bands, it is recommended to try a different PCR reaction system.
- Nested PCR reaction: A nested or semi-nested PCR method is advisable to obtain sufficient PCR products, especially in the case of small amounts of DNA.
Note: The PCR results will be verified by gel-based electrophoresis and a single, bright and specific band will be considered as a successful PCR amplification.
Direct PCR Sequencing and Cloning Sequencing
- Prior to the direct PCR sequencing, purification of PCR products is necessary to remove the residue of the PCR reaction that might interfere with the results of sequencing outcomes. Commercially-available kits such as PCR Purification Kit can be used for specific PCR fragments, whereas Gel Extraction Kit can help purify the target PCR product from multiple nonspecific PCR bands. The purified PCR products can be directly sequenced.
- Cloning sequencing is necessary to observe the distribution of methylation patterns in single molecules. We prefer to use pGEM-T Easy Vector System II for cloning purposes which provides the T4 DNA ligase system, a pGEM-T Easy vector and competent JM109 cells as well. By using this kit, purified PCR products can be ligated to the pGEM-T Easy vector and transformed into competent JM109 cells. The JM109 cells that carry the ligated vectors can be selected on agar plates containing ampicillin/X-gal/IPTG by color change where blue colonies represent empty vector, and white colonies represent vectors inserted with target PCR product. The white colonies can then be selected and grown in LB medium. Plasmids containing the target DNA are extracted by using the Spin Miniprep Kit and subjected to standard sequencing analysis. All the procedures follow the manufacturer’s protocol.
References
- Ino D. Karemaker, et al.; Single-cell DNA methylation profiling: technologies and biological applications. Trends in Biotechnology, 36(9): 952-965.
- Zuo T, et al.; Methods in DNA methylation profiling. Epigenomics, 2009, 1(2): 331-345.
Cell Services:
Cell Line Testing and Assays