Inhibition of Lin28 Restores Lipid Homeostasis in Disease Models
Nature Communications. 2022 Dec 26; 13(1): 7940
Authors: Lekka E, Kokanovic A, Mosole S, Civenni G, Schmidli S, Laski A, Ghidini A, Iyer P, Berk C, Behera A, Catapano CV, Hall J.
INTRODUCTION
- Lin28 RNA-binding proteins are stem-cell factors that play key roles in development. Lin28 suppresses the biogenesis of let-7 microRNAs and regulates mRNA translation. Notably, let-7 inhibits Lin28, establishing a double-negative feedback loop. The Lin28 / let-7 axis resides at the interface of metabolic reprogramming and oncogenesis and is therefore a potential target for several diseases.
- Small-molecule ligands offer a direct means to identify disease areas in which a new target can be effectively and safely exploited. The search for small-molecule inhibitors of the Lin28 / let-7 interaction began shortly after the RNA-protein complex was characterized. We identified the small molecule C1632 from an innovative high-throughput screen of 16000 drug-like compounds.
METHODS
- Recently, we developed and used an innovative high-throughput screening assay to identify drug-like repurposing compounds, including two unrelated independent structures (C1632, C4019) that inhibit the Lin28 / let-7 interaction and restore the processing and function of let-7 miRNAs. To examine the effects of C1632 in vivo, we administered it (50 mg / kg) by intraperitoneal injections on 5 consecutive days to 7-week-old, male C57BL / 6J wild-type mice feeding on a normal diet.
- We examined the expression of known regulators of ketogenesis in livers of vehicle- and C1632-treated mice, and the mRNA levels of PPARα and its downstream gene targets from the pathway. Then, we investigated whether compound C1632 could control lipid homeostasis and block oleic acid-induced lipid accumulation in two commonly used cellular models of hepatic steatosis in murine AML12 hepatocytes and human HepG2 cells.
- To investigate whether promoting ketogenesis and fatty acid catabolism via pharmacological inhibition of Lin28 / let-7 could be beneficial in liver disease, we turned to established mouse models of non-alcoholic fatty liver disease (NAFLD). To determine whether pharmacological Lin28 inhibition could retard the development of liver steatosis, we initially treated male Pten-/- mice at early age (6 weeks old) with C1632 or vehicle for 4 weeks, then, we repeated the C1632 treatment in 22 weeks-old Pten-/- male mice with three injections of C1632 per week for six weeks.
RESULTS
- We observed mildly increased blood glucose levels in treated mice compared with controls. Lin28A and Lin28B were reduced by approximately 20% and 40%, respectively, in liver, and by approximately 30% and 60% in skeletal muscle, consistent with a broad distribution and activity of C1632 in vivo. AKT Ser473 phosphorylation (p-AKT) was reduced in the liver and skeletal muscle from C1632-treated mice over the control group.
- Compared to controls, serum levels of β-hydroxybutyrate (β-OHB), the most abundant ketone body54, were significantly higher in C1632-treated mice. In liver, NCoR1 inhibits PPARα activation and its downstream targets, attenuating ketogenesis. Consistent with this role, we found that levels of the parent NCoR1 protein in the livers of C1632-treated 8-week-old male wild-type mice were reduced by approximately two-fold. Furthermore, C1632 treatment lowered levels of NCoR1 in HepG2 cells.
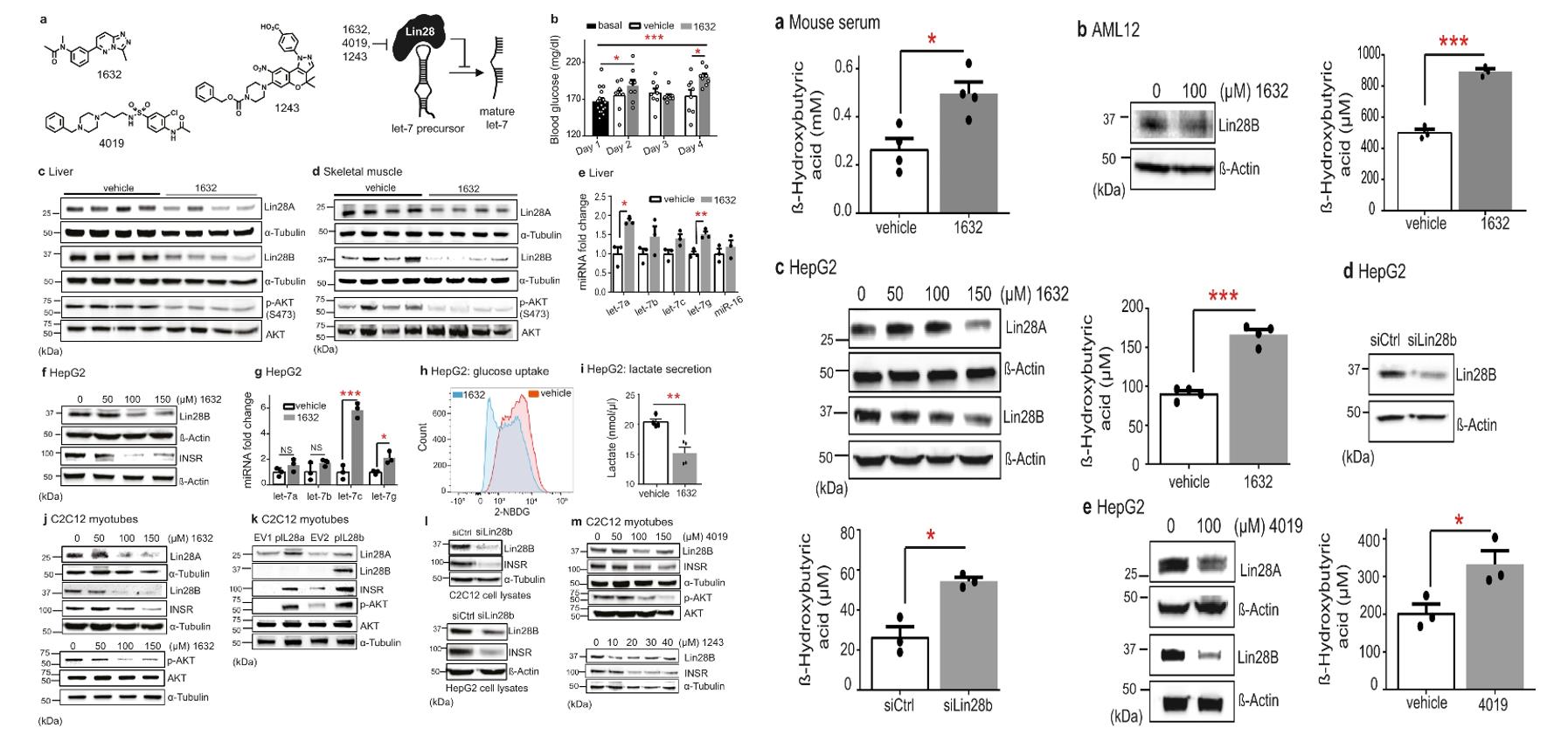 Fig. 1 Left: Lin28A / B inhibition suppresses insulin-PI3K-mTOR signaling in liver and skeletal muscle; Right: The small molecule Lin28 inhibitor C1632 promotes ketogenesis.
Fig. 1 Left: Lin28A / B inhibition suppresses insulin-PI3K-mTOR signaling in liver and skeletal muscle; Right: The small molecule Lin28 inhibitor C1632 promotes ketogenesis.
- Exposure of the non-tumorigenic mouse hepatocyte cells AML12 to oleic acid promoted intracellular lipid accumulation, which was alleviated by C1632 treatment, as shown by Oil Red O staining. Similarly, in basal conditions, HepG2 cells show low levels of lipid deposits. And the expression of enzymes involved in fatty acid and triglyceride synthesis were decreased, whereas those involved in mitochondrial β-oxidation and ketogenesis increased.
- In the short-term experiments, levels of Lin28A / B and NCoR1 proteins were decreased, and similar changes were observed for lipogenesis- and ketogenesis-related mRNAs, as well as serum β-OHB levels. From C1632-treated mice now revealed an overall decline in the mean size of lipid droplets and a decrease in the number and size of large lipid droplets. This was consistent with reduced macro vesicular steatosis in the hepatocytes.
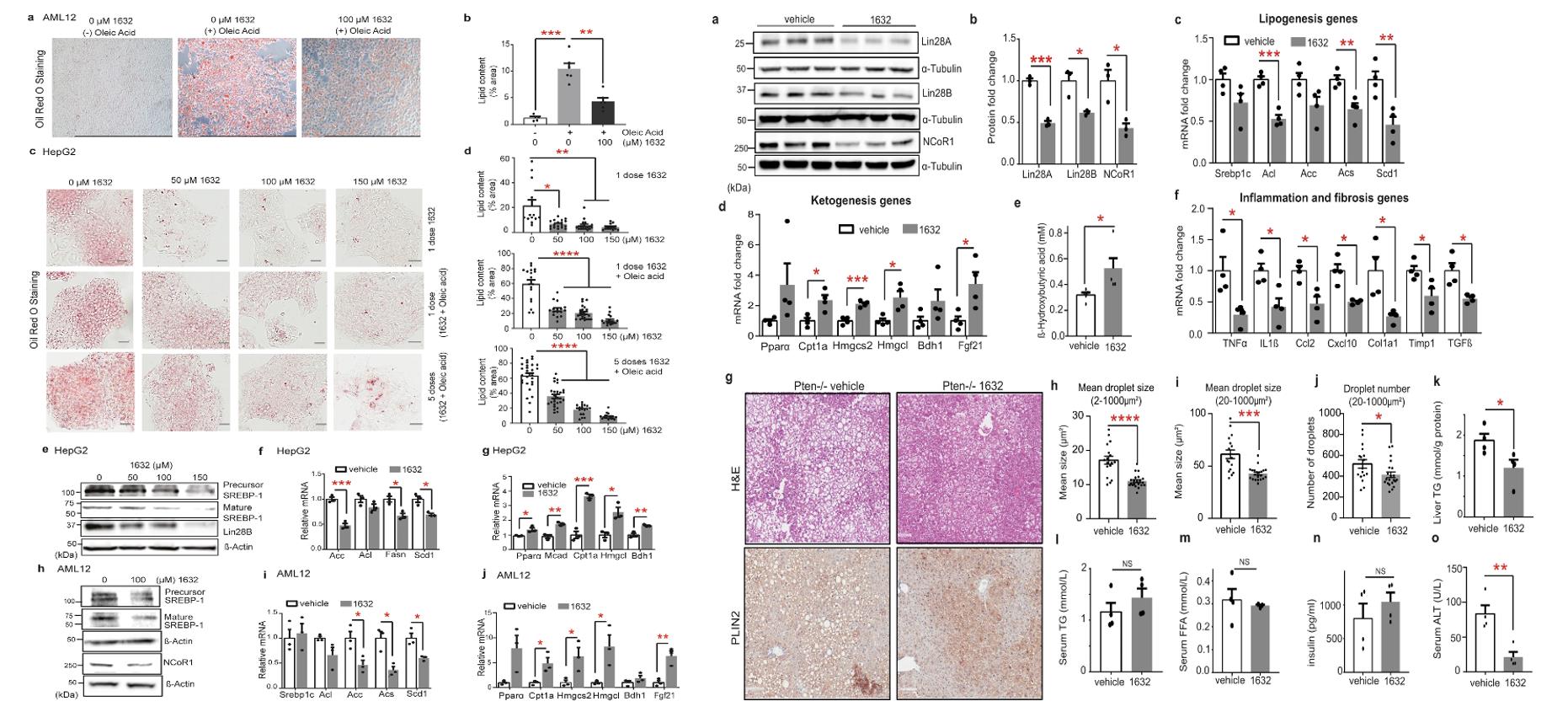 Fig. 2 Left: C1632 improves lipid metabolism in hepatocytes in vitro; Right: C1632 attenuates hepatosteatosis in a genetic mouse model of NAFLD.
Fig. 2 Left: C1632 improves lipid metabolism in hepatocytes in vitro; Right: C1632 attenuates hepatosteatosis in a genetic mouse model of NAFLD.
SUMMARY
- Lin28 inhibition enhances lipid catabolism and ketogenesis, whilst repressing SREBP1-mediated anabolic lipogenesis, which together reduces intracellular lipid accumulation in HepG2 and AML12 cells.
- Finally, C1632 treatment activates an antisteatotic program in both genetic and dietary mouse models of non-alcoholic fatty liver disease (NAFLD), suggesting that a moderate pharmacological inhibition of Lin28 might be beneficial for the prevention or treatment of NAFLD.
RELATED PRODUCTS & SERVICES
Reference
- Lekka E, et al. (2022). "Pharmacological inhibition of Lin28 promotes ketogenesis and restores lipid homeostasis in models of non-alcoholic fatty liver disease." Nat Commun. 13 (1), 7940.