Analysis of Molecular Classification and Biomarkers in Breast Ductal Carcinoma in Situ
Cancer Cell. 2022 Dec 12; 40(12): 1521-1536
Authors: Strand SH, Rivero-Gutiérrez B, Houlahan KE, Seoane JA, King LM, Risom T, Simpson LA, Vennam S, Khan A, Cisneros L, Hardman T, Harmon B, Couch F, Gallagher K, Kilgore M, Wei S, DeMichele A, King T, McAuliffe PF, Nangia J, Lee J, Tseng J, Storniolo AM, Thompson AM, Gupta GP, Burns R, Veis DJ, DeSchryver K, Zhu C, Matusiak M, Wang J, Zhu SX, Tappenden J, Ding DY, Zhang D, Luo J, Jiang S, Varma S, Anderson L, Straub C, Srivastava S, Curtis C, Tibshirani R, Angelo RM, Hall A, Owzar K, Polyak K, Maley C, Marks JR, Colditz GA, Hwang ES, West RB.
INTRODUCTION
- Breast ductal carcinoma in situ (DCIS) is one of the most common precancers across all tissues, with 50,000 women diagnosed each year in the United States. DCIS is the most common precursor of invasive breast cancer (IBC), with variable propensity for progression. Previous molecular analyses of DCIS have studied either cohorts of pure DCIS with known outcomes (e.g., disease-free vs. recurrent) or cross-sectional cohorts of DCIS with or without adjacent IBC.
- Here, as part of the Human Tumor Atlas Network (HTAN), the study presents two DCIS cohorts, the Translational Breast Cancer Research Consortium (TBCRC) 038 study and the Resource of Archival Breast Tissue (RAHBT), for multimodal molecular analyses. Performing comprehensive integrated molecular profiling of these complementary, clinically annotated, longitudinally sampled cohorts to understand the spectrum of molecular changes in DCIS and to identify both tumor and stromal predictors of subsequent events.
METHODS
- The researchers generated two retrospective study cohorts, consisting of DCIS patients with either a subsequent ipsilateral breast event (iBE) or no later events after surgical treatment. TBCRC samples were macro dissected for downstream RNA and DNA analyses. RAHBT samples were macro dissected like TBCRC or organized into a tissue microarray (TMA) from which serial sections were made for RNA, DNA, and protein (MIBI) analysis (RAHBT LCM cohort).
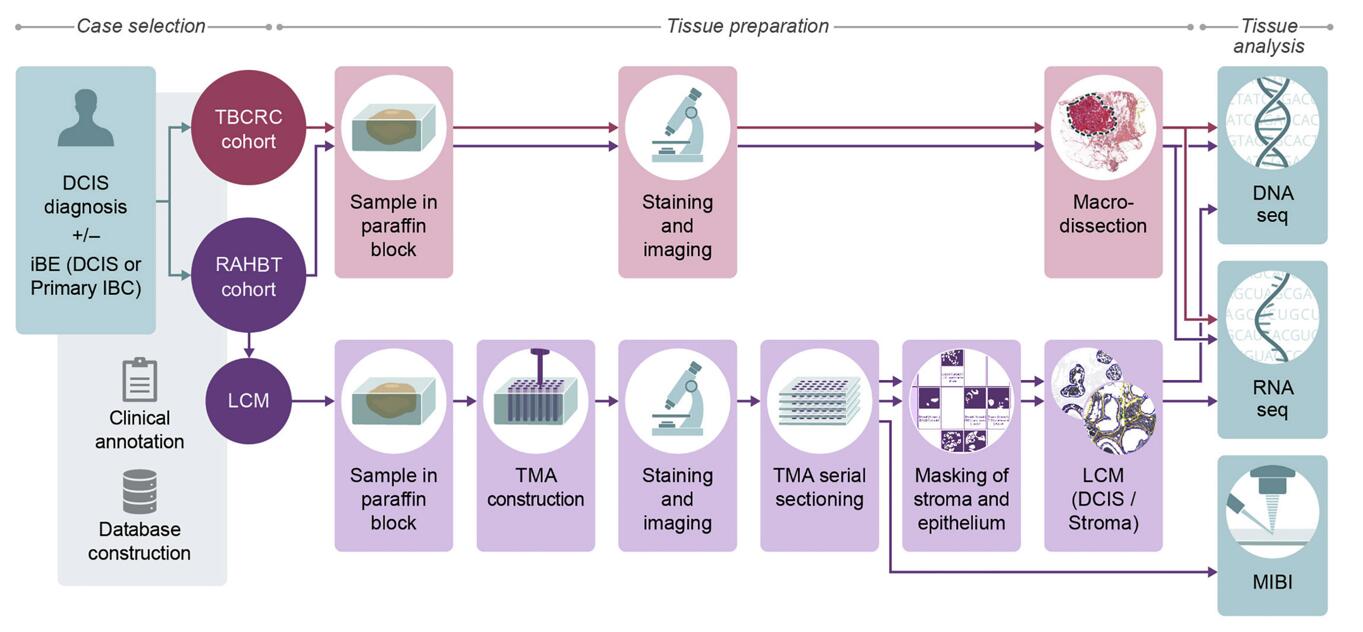 Fig. 1 Cohorts and methods outline.
Fig. 1 Cohorts and methods outline.
- To identify gene expression patterns correlating with outcome, the researchers analyzed RNA from primary DCIS with iBEs within 5 years vs. the remaining samples in TBCRC, then, the study trained a random forest classifier in TBCRC using these differentially expressed (DE) genes. To further examine pathway activation status, we performed gene set variation analysis (GSVA) at the individual tumor level in 5-year outcome groups.
- The study performed non-negative matrix factorization (NMF) on all protein coding genes (GENCODE v33) with non-zero variance, evaluated the fit of 2-10 clusters and selected a three-cluster solution. In support of the three-cluster solution, we investigated MIBI protein expression for a subset of patients (n = 71). To identify enriched pathways in the eight CNA clusters, we investigated the differential abundance in matched RNA samples (DESeq2 one-vs.-rest) and performed GSEA hallmark analysis on the resulting gene lists.
- Using adjacent TMA sections to analyze RNA and MIBI expression on the same ducts. The researchers compared MIBI-based cell type distribution across samples with the inferred cell type distribution from RNA expression data using CIBERSORTx, allowing to cross-validate findings and extend observations on cell composition to DCIS samples without MIBI data, including the TBCRC cohort.
RESULTS
- Given the absence of significant CNAs, we trained a random forest classifier in TBCRC using only the 812 DE genes. The classifier was validated in RAHBT, indicating that the classifier performed well also in the test cohort. Importantly it was also a significant predictor of invasive iBEs over the full follow-up time, demonstrating the classifier could specifically identify DCIS that progress to IBC. In the study, MYC and mTORc1 signaling were increased in cases vs. controls and strongly correlated.
- In both cohorts, cluster 1 had significantly higher ERBB2 and lower ESR1 expression compared with clusters 2 and 3, which both had increased ESR1 expression. We termed the three clusters ERlow, quiescent, and ERhigh respectively. Based on MIBI data, quiescent lesions were depleted for Ki67 and GLUT1 positive tumor cells, vs. ERhigh and ERlow tumors. ERhigh lesions had significantly higher myoepithelial E-cadherin frequency compared with ERlow and quiescent lesions. While most recurrence-associated pathways were enriched in ERlow lesions, this points to a feature associated with recurrence among ER+ DCIS tumors, and it highlights that there are multiple paths to progression in DCIS.
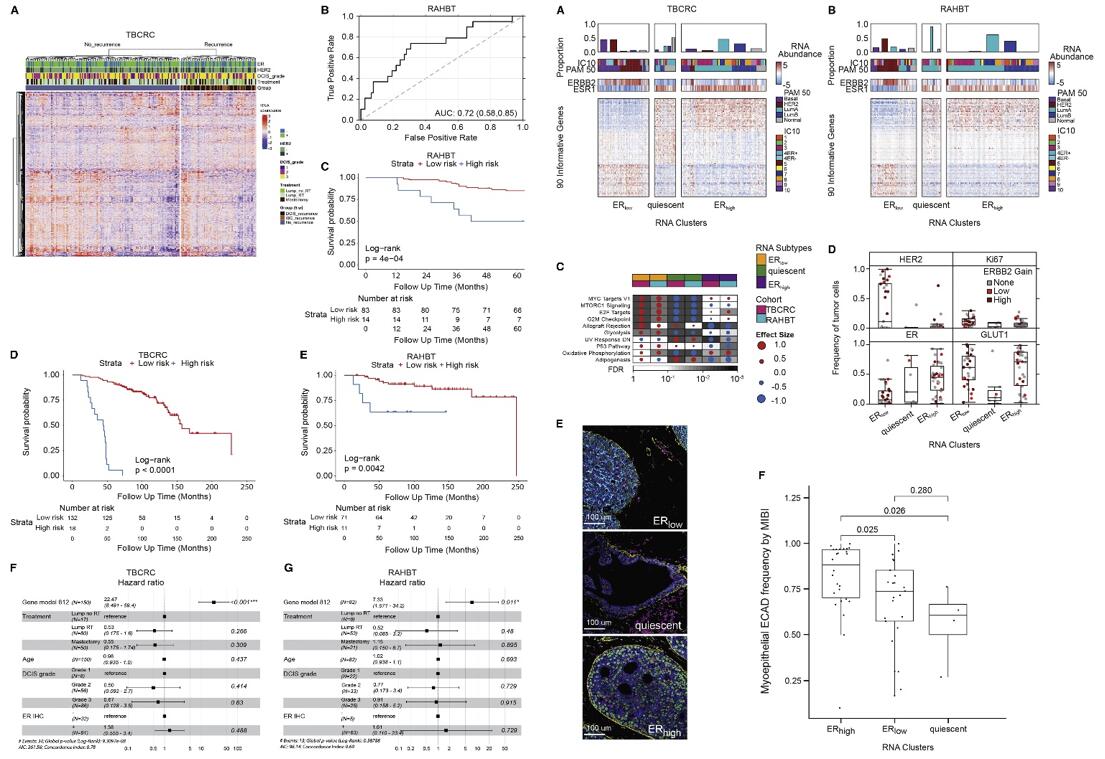 Fig. 2 Left: Identification, training, and validation of 812-gene classifier; Right: Transcriptomic DCIS subtypes correlate with outcome pathways.
Fig. 2 Left: Identification, training, and validation of 812-gene classifier; Right: Transcriptomic DCIS subtypes correlate with outcome pathways.
- Clusters 6 and 7 were enriched for pathways associated with recurrence (allograft rejection and oxidative phosphorylation, respectively), whereas cluster 8 was depleted of recurrence-associated pathways (cell cycle and mTORc1 signaling), and cluster 6 was depleted of MYC targets. Thus, we identified a CNA-based cluster solution characterized by amplifications seen in high-risk IBC subtypes, including 17q12 (ERBB2) and 8q24 (MYC) amplification, some of which were significantly enriched or depleted for pathways associated with recurrence.
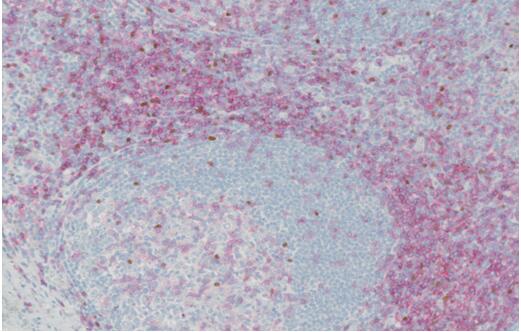
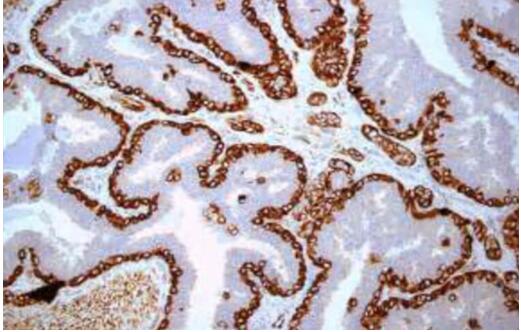
- We performed shared nearest neighborhood clustering of stromal RNA data and identified four distinct DCIS-associated stromal clusters and DE genes. Pathway analyses, MIBI protein expression and cell type distribution, and CSx-inferred cell type distribution were used to describe major characteristics of each cluster, which were termed "immune dense," "desmoplastic," "collagen-rich," and "normal-like." Representative MIBI images of each cluster are correlated with fibroblast states and immune cell density.
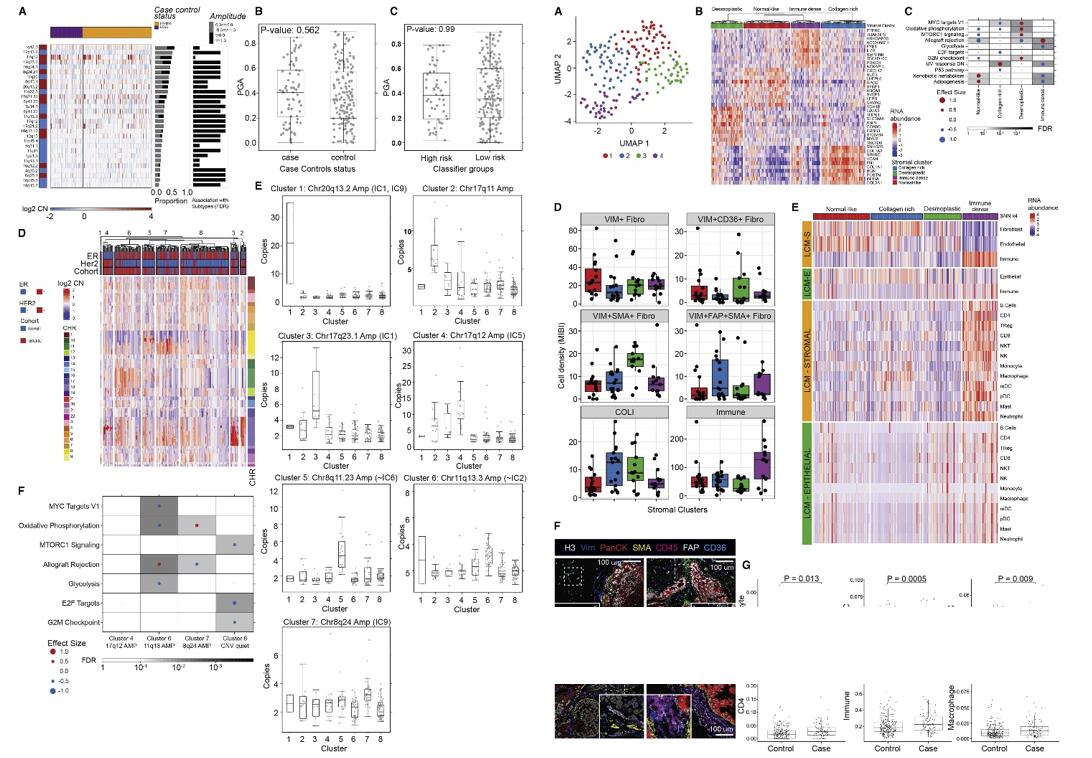 Fig. 3 Left: Characteristic IBC CNAs are present in DCIS; Right: TME analysis.
Fig. 3 Left: Characteristic IBC CNAs are present in DCIS; Right: TME analysis.
SUMMARY
- This study performs multiscale, integrated molecular profiling of DCIS with clinical outcomes by analyzing 774 DCIS samples from the Translational Breast Cancer Research Consortium 038 study and the Resource of Archival Breast Tissue cohorts. Identifying 812 genes associated with ipsilateral recurrence within 5 years from treatment and develop a classifier that predicts DCIS or IBC recurrence in both cohorts.
- Pathways associated with recurrence include proliferation, immune response, and metabolism. Distinct stromal expression patterns and immune cell compositions are identified.
- There is multiscale approach employed in situ methods to generate a spatially resolved atlas of breast precancers, where complementary modalities can be directly compared and correlated with conventional pathology findings, disease states, and clinical outcome.
RELATED PRODUCTS & SERVICES
Reference
- Strand SH, et al. (2022). "Molecular classification and biomarkers of clinical outcome in breast ductal carcinoma in situ: Analysis of TBCRC 038 and RAHBT cohorts." Cancer Cell. S1535-6108 (22), 00512-8.